Cytochrome P450 Time Dependent Inhibition (kinact/KI) Assay
Understand the potential drug-drug interaction liabilities of your compounds by using our cytochrome P450 (CYP450) time dependent inhibition assays for a range of isoforms.
Our range of cytochrome P450 time dependent inhibition services form part of our portfolio of in vitro ADME services. Cyprotex delivers consistent, high quality data with the flexibility to adapt protocols based on specific customer requirements.
Introduction
Time dependent inhibition of cytochrome P450 (CYP450) enzymes:
- Time dependent inhibition of cytochrome P450, often caused by an irreversible or quasi-irreversible interaction, can lead to clinically relevant drug-drug interactions or non-linear pharmacokinetics of a drug. In addition, these interactions are typically a consequence of reactive metabolite formation which is also associated with toxicity via covalent binding to cellular macromolecules1.
- Characterisation of the kinact (maximal inactivation) and KI (concentration at 50% kinact) parameters is frequently performed during drug development to evaluate risk of time dependent inhibition and decide if a clinical interaction study is required.
- Cyprotex’s kinact/KI assay evaluates the inactivation kinetics of time dependent inhibition at 5 inhibitor concentrations and 7 pre-incubation times.
Related Services:
Protocol
Cytochrome P450 Time Dependent Inhibition (kinact/KI) Assay Protocol
Data
Data from the Cytochrome P450 Time Dependent Inhibition (kinact/KI) Assay
A number of known time dependent inhibitors were characterised in the kinact/KI assay and compared with data published in the literature.
Q&A
Please provide an overview of the Cytochrome P450 time dependent inhibition: kinact/KI.
The Cytochrome P450 Time Dependent Inhibition: kinact/KI assay determines kinetic constants of time dependent inhibition. The kinact is the maximal rate of enzyme inactivation at a saturating concentration of inhibitor and KI is the concentration of inhibitor which gives half the maximal rate of inactivation. The experimental conditions are determined from the results of previously performed assays such as P450 reversible inhibition and time-dependent inhibition: single point or IC50 shift. Compounds are evaluated by pre-incubating a range of 7 test compound concentrations (plus a vehicle control) selected so to achieve a scale from no inactivation to maximal inactivation, for 6 differing pre-incubation times (including 0 min) with human liver microsomes and NADPH. Following the pre-incubation, an aliquot of the pre-incubation is diluted with buffer containing a specific cytochrome P450 probe substrate (at 5×Km concentration) and NADPH for a substrate specific incubation time. Following least-squares regression analysis to determine the negative slope of the logarithm of the % remaining activity (corrected for negative control) versus pre-incubation time, non-linear regression analysis of the negative slopes against inhibitor concentration enables kinact and KI to be calculated (Figure 3).
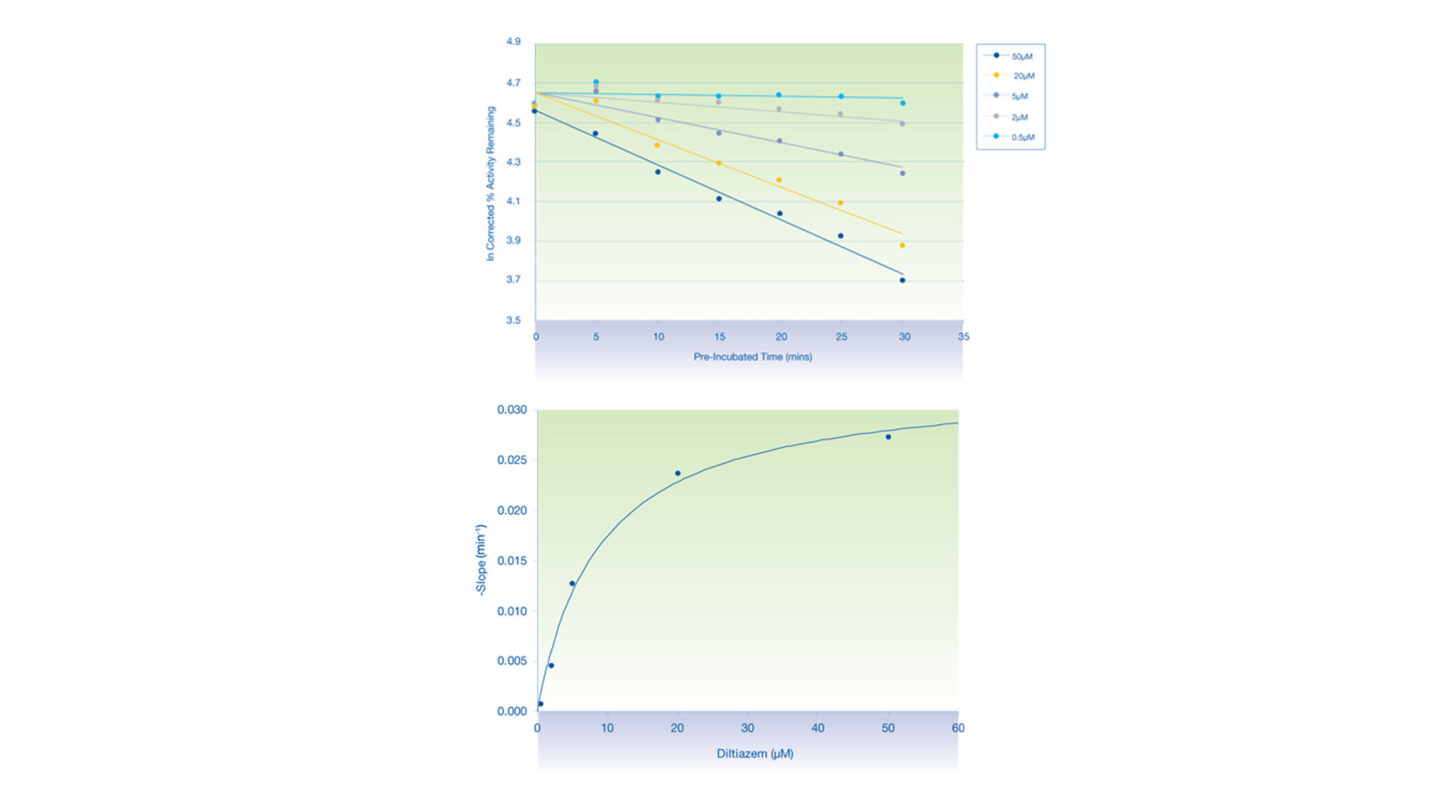
Figure 3
The inactivation plots of CYP3A4 by diltiazem:
- (Above) Plot of natural logarithm of the corrected % activity remaining against pre-incubation time.
- (Below) Non-linear regression analysis of the observed rates of inactivation against inhibitor concentration.
- Each data point represents the mean of n=2.
Why is it important to investigate time dependent inhibition?
Cytochromes P450 (CYP) are a family of enzymes which play a major role in the metabolism of drugs. Inhibition of CYP enzymes is one of the most common causes of drug-drug interactions. The mechanism of inhibition can be reversible, quasi-irreversible or irreversible.
The consequences of irreversible inhibition are considered to be more serious than reversible inhibition because the inactivated enzyme must be re-synthesized before activity is restored. In addition, the irreversible inactivation usually implies the formation of a covalent bond between the metabolite and the enzyme, which can lead to hapten formation and can in some cases trigger an autoimmune-response. For these reasons it is important to study the mechanism of the CYP inhibition of new potential drugs as early as possible during the drug discovery process.
Although the terms time dependent inhibition (TDI) and mechanism based inhibition (MBI) are often used inter-changeably, there is a distinct difference scientifically. Time dependent inhibition is defined as an interaction where there is an enhanced inhibition if the test compound is pre-incubated with the metabolizing system prior to addition of the substrate. Mechanism based inhibition specifically refers to a subset of time dependent inhibition which defines inactivation of the enzyme by a chemically reactive metabolite2.
The FDA guidance for drug interactions (2020)3 and the EMA guideline on the investigation of drug interactions (adopted 2012)8 recommend evaluating time dependent inhibition for investigational drugs.
What information is needed prior to performing a kinact/KI determination?
The kinact/KI determination assay is a later stage assessment of inhibitor kinetics. The data analysis and utility of the data from this assay is completely dependent upon the choice of inhibitor concentrations and pre-incubation times. Ideally the combination of both of these variables should be so that a range of inactivation is achieved from maximal inactivation to no inactivation.
This can be difficult to achieve if the compound undergoes considerable reversible inhibition during the substrate incubation. Therefore it is important to have knowledge of the potency of reversible inhibition beforehand so suitable conditions can be determined e.g. a greater than 1:10 dilution to dilute the test compound before substrate incubation, or increased pre-incubation time compared to incubation time. An indication of the amount of time-dependent inhibition expected is also required, from either a single point or IC50 shift assay. This provides an indication of the highest inhibitor concentration required to give maximal inhibition.
How are the results from the kinact/KI assay interpreted?
The kinact and KI values determined can be used to predict the risk of drug-drug interactions. The FDA guidance for drug interactions (Jan 2020)3 recommend determining kinact and KI if any time dependent loss of initial product formation rate is observed in preliminary in vitro pre-incubation studies. An estimated R value of greater than 1.1 where R= (Kobs+ Kdeg)/Kdeg and Kobs = kinact x [I]/(KI +[I]) is considered positive and thus may warrant further in vivo investigations. [I] represents the maximal total (free and bound) systemic inhibitor concentration in plasma. For CYP3A4 inhibitors that are orally dosed, [I] should also be estimated by [I] = Igut = molar dose/250mL and if the alternate R is greater than 11 then further in vivo investigations may be warranted. Mechanistic and dynamic models may also be used to help make the decision on whether to proceed to in vivo studies.
Within the EMA guideline on the investigation of drug interactions (adpoted 2012)8, a multiple dose in vivo interaction study is recommended if greater than or equal to 20% inhibition is observed (i.e. R ≥ 1.25). The suggested approach is to calculate the ratio of predicted clearance in the absence and presence of the inhibitor using the following equation;
R = (Kobs + kdeg)/kdeg and Kobs = kinact × [I]/(KI + [I])
Where kdeg is the degradation constant of the enzyme, kinact is the maximum inactivation constant and [I] is the concentration of inhibitor2.
References
1) Kalgutkar AS et al. (2007) Curr Drug Metab 8; 407-447
2) Grimm SW et al. (2009) Drug Metab Dispos 37; 1355-1370
3) FDA Guidance for Industry – In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions (January 2020)
4) Obach RS et al. (2007) Drug Metab Dispos 35(2); 246-255
5) Prueksaritanont T et al. (1999) Br J Clin Pharmacol 47; 291-298
6) Berry LM et al. (2008) Drug Metab Lett 2; 51-59
7) Perloff ES et al. (2009) Xenobiotica 39(2); 99-112
8) The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions (Adopted in 2012)
Downloads
- Cyprotex ADME Guide 5th Edition >
- Cyprotex DDI Regulatory Guide 3rd Edition >
- Cyprotex Time Dependent CYP Inhibition (kinact/KI) Factsheet >