Cytochrome (CYP) P450 Inhibition Ki Assay
Use the cytochrome P450 (CYP) inhibition Ki assay to understand the relevance and type of reversible cytochrome P450 inhibition.
Cytochrome P450 inhibition Ki determination is one of our portfolio of in vitro experimental ADME services. Cyprotex delivers consistent, high quality data with the flexibility to adapt protocols based on specific customer requirements.
Introduction
Determining the clinical relevance of reversible cytochrome P450 (CYP) inhibitors:
- Assessment of the potential of a compound to inhibit a specific cytochrome P450 (CYP) enzyme is important as co-administration of compounds may result in one or both inhibiting the other’s metabolism. This may affect plasma levels in vivo and potentially lead to adverse drug reactions or toxicity.
- Determination of the inhibition constant (Ki) of a compound is the current recommended approach by the FDA for studying the clinical relevance of reversible cytochrome P450 inhibitors.
- Cyprotex's cytochrome P450 Inhibition Ki assay delivers a written report detailing graphical representation of the data and calculation of the Ki value. The type of inhibition is determined by fitting statistics for the enzyme inhibition models (i.e. competitive, non-competitive, uncompetitive and mixed).
Protocol
Cytochrome P450 (CYP) Inhibition Ki Assay Protocol
Data
Data from Cyprotex's Cytochrome P450 (CYP) Inhibition Ki assay
Q&A
Why is it important to investigate cytochrome P450 inhibition?
Cytochromes P450 (CYP) are a family of enzymes which play a major role in the metabolism of drugs. Assessment of the potential of a compound to inhibit a specific cytochrome P450 enzyme is important as co-administration of compounds may result in one or both inhibiting the other’s metabolism. This may affect plasma levels in vivo and potentially lead to adverse drug reactions or toxicity. Determination of the inhibition constant (Ki) of a compound is the current recommended approach by the FDA and EMA for studying the clinical relevance of reversible cytochrome P450 inhibitors.
Please provide an overview of Cyprotex's Cytochrome P450 (CYP) Inhibition Ki assay.
Compounds are evaluated incubating up to 6 inhibitor concentrations, (spanning the IC50 determined using Cyprotex's Cytochrome P450 Inhibition IC50 assay), and 5 substrate concentrations, spanning the Km, in duplicate. Determination of the Ki value of a test compound defines the inhibitory affinity of the compound for a particular enzyme and can be used to estimate the potential clinical significance of in vitro inhibition.
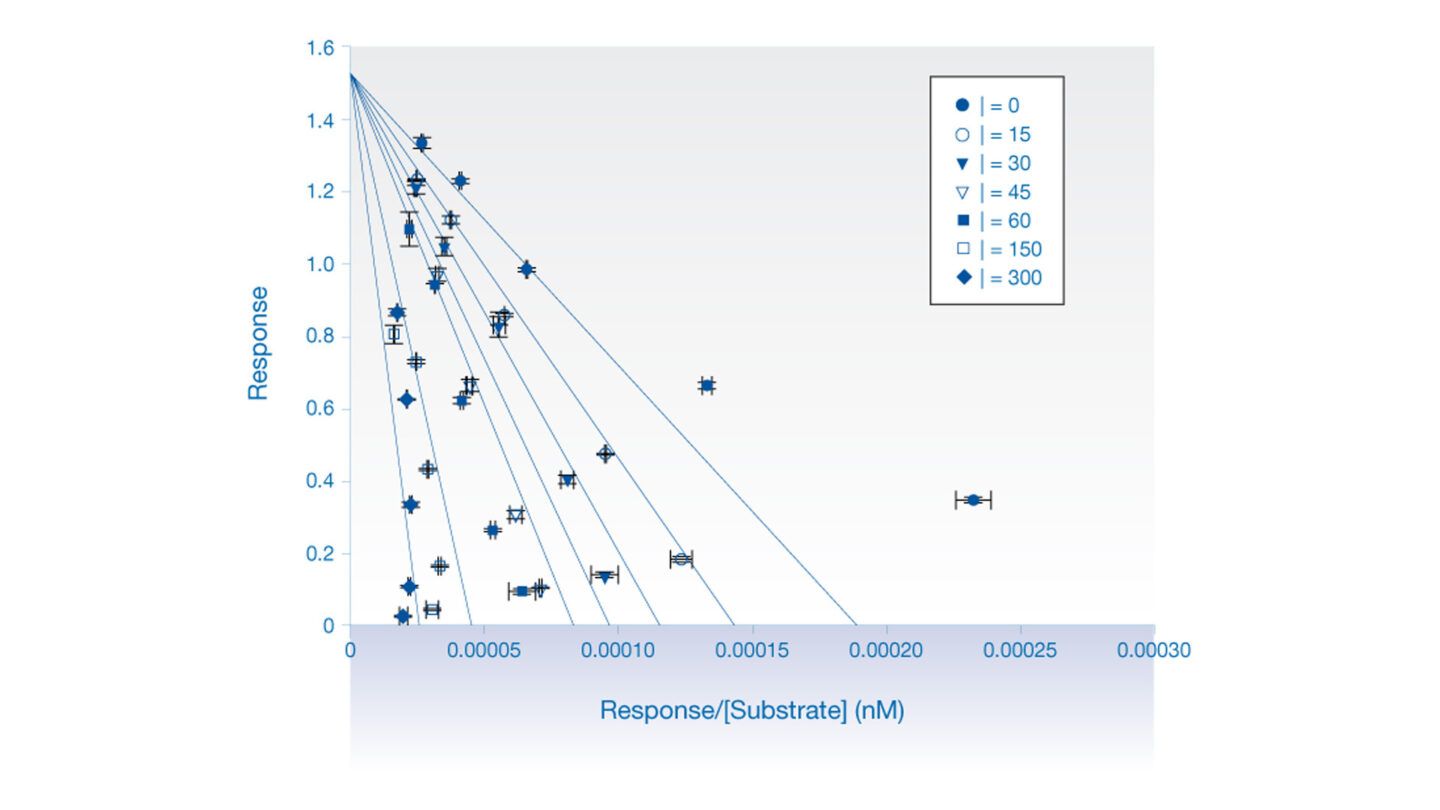
Figure 1
Eadie-Hofstee (competitive inhibition) analysis of the effect of quinidine on dextrorphan formation in human liver microsomes.
Each data point represents the mean ± SE (n=2). [I] = nM.
What is the difference between IC50 and Ki?
Ki is an intrinsic inhibitor constant which defines the affinity with which an inhibitor binds to an enzyme. The IC50 of an enzyme is the concentration of inhibitor which produces 50% inhibition. Preliminary IC50 determination provides a guide to the inhibitor concentrations to be used in a Ki determination assay. The IC50 value of a test compound is dependent upon experimental conditions such as the substrate concentration, incubation time and protein concentration, and therefore may not be reproducible from lab to lab. Ki is characteristic for each particular inhibitor and enzyme, and is not dependent on any particular substrate used. The Ki value is a more reproducible and more reliable value to give an indication whether future in vivo work is required.
How is the best-fit model chosen?
Regression analysis is used to identify the type of inhibition (competitive, non-competitive, uncompetitive or mixed) and the most appropriate inhibition model selected based upon the goodness of fit criteria of a visual inspection of the data, correlation of determination (R2) and corrected Akaike’s Information Criterion (AICc). For visual inspection, data are presented as a direct plot of response against substrate concentration, in addition to Eadie-Hofstee and Lineweaver-Burk plots.
What does alpha mean?
Alpha (α) is the interaction parameter applied to the mixed inhibition model, which determines the degree to which the binding of inhibitor changes the affinity of the enzyme for the substrate. For example, when α = 1, the inhibitor does not change the binding of the substrate to the enzyme, thus making the mixed inhibition model identical to the non-competitive model. When α > 1 this means binding of the inhibitor is preventing binding of the substrate, reducing the formation of an enzyme-substrate-inhibitor complex, thus pushing the model towards a competitive inhibition model. When α < 1, binding of inhibitor enhances binding of substrate, pushing the model towards an uncompetitive inhibition model.
How do I interpret the data and utilise the Ki value?
For reversible inhibition, a simple classification, based on the [I]/Ki ratio (where [I] is the maximal total (free and bound) systemic inhibitor concentration in plasma), is commonly used to predict clinical drug-drug interactions. As per FDA guidelines, an estimated R = 1+[I]/Ki of greater than 1.02 is considered positive and thus may warrant further in vivo investigations. For CYP3A4 inhibitors that are orally dosed, [I] should also be estimated by [I] = Igut = molar dose/250mL and if the alternate R is greater than 11 then further in vivo investigations may be warranted. Mechanistic or dynamic models may also be used to help make the decision on whether to proceed to in vivo studies.
For the EMA guideline on the investigation of drug interactions (adopted 2012)5, if the enzyme studied is mainly in the liver, or the kidney / another organ with main drug input from the systemic circulation, then an in vivo interaction study with a sensitive probe substrate is recommended if [I]/Ki ≥ 0.02 where [I] is the unbound mean Cmax obtained during treatment with the highest recommended dose. For orally administered drugs if the enzyme has marked abundance in the enterocyte (e.g., CYP3A), an in vivo study is recommended if [I]/Ki ≥ 10 where [I] is the maximum dose taken at one occasion/250mL. In these calculations for the EMA guideline the lowest figure for free fraction recommended is 1% due to the uncertainties in the estimation. Mechanistic or dynamic (PBPK) models may also be used for decision making on clinical relevance of the in vitro data.
References
1) Ogilvie BW et al. (2008) in Drug-Drug Interactions Second Edition (Ed. Rodrigues AD) Informa Healthcare USA New York 231-358
2) Wrighton SA and Ring BJ. (1994) Pharmaceutical Research 11 (6); 921-924
3) Gibbs MA et al. (1999) Drug Metab Dispos 27(2); 180-187
4) Brown HS et al. (2007) Drug Metab Dispos 35(11); 2119-2126
5) The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions (Adopted in 2012)
Downloads
- Cyprotex ADME Guide 5th Edition >
- Cyprotex DDI Regulatory Guide 3rd Edition >
- Cyprotex CYP Inhibition Ki Assay Factsheet >